We are kicking off the alpha of our new platform for building bioinformatics applications!
We at Genestack are dedicated to making the life of a bioinformatician, whether embedded in a hospital, running the bioinformatics for a biotech, working in a computational biology unit in a pharma company, or just trying to conduct research in an organisation without a significant IT/systems/bioinformatics infrastructure and support. We built a platform that allows users to store and share large data sets securely within and across organisations, with free access to public data from major databases. The platform includes open-source and proprietary genomics applications, working together independent of file formats. We provide an SDK for writing interactive, visual and computational applications in several languages.
We are now inviting users to try out a public alpha of the platform. In it you will have a chance to explore our data management system and try some of the first apps we've written. We'll roll out frequent updates to the system and let you know about the changes right here.
Key Concepts
First we'd like to highlight some key concepts unique to our platform. Everything is a File Almost all objects that you will see in Genestack Platform are files. The output from a sequencer is a file (Sequencing assay). Sequenced reads that are aligned and mapped against a reference genome are a file (Aligned and mapped reads). An Affymetrix CEL file from a microarray assay is a file, too, of course, as you'd expect it to be (Microarray assay). But so is a reference genome, a file (Reference genome). A genome browser page is a file. An experiment from Gene Expression Omnibus is a file in our system, too. It is an extremely useful metaphor, as you will see, once you start using the system. Files in Genestack Platform have rich metadata, and are actually data packages with functionality. Files expose useful interfaces, natural to the data that files contain. Thus, Reference genome contains methods to query for genome features, while a Sequencing assay allows retrieving individual sequence reads, with qualities, if available. This is quite useful for programmers. A File is More Than a File Files in Genestack Platform are not the same files as those you will find on your machine, even though we call them by the same name. For example, a .fastq file contains short reads from a sequencer with qualities. But so does a .sra file from the Short Read Archive, but in a different format. Our files are format-free biologically meaningful objects: a set of sequences, or a reference genome. When working with these files you are guaranteed that they contain the right information, and you do not have to worry what format that information is stored in. The platform will automatically take care of any conversions, adaptations as necessary. Moreover, metadata attributes record where each file came from, how it was generated, and so forth. Metadata fields are typed, meaning that if a field is supposed to be numeric, it will necessarily contain a number, and if a field is supposed to be a publication with a PubMed ID, it will definitely have such an ID. This means that both users and the system can impose some consistency constraints on the data. Files Everywhere A special feature of our file system is that one file can be present in several folders. This is similar to hard links in Unix. An immediate advantage of this approach becomes apparent when dealing with files of very large sizes, as is common in next generation sequencing workflows: you will never need to duplicate your files anymore. A copy operation becomes cheap in Genestack Platform. Similarly, sharing data is natural and easy. Lazy Initialisation (Tasks) Because our files are not quite the same as files in the ordinary sense, we can do some fancy tricks with them. One of these tricks is that a file can serve as a mere placeholder for the data, waiting to be initialised until the time someone actually needs it. It might have all (or some) of the metadata attributes filled in, and when you first use it, the data and all metadata fields will be updated. For example, the file for a reference genome for an infrequently accessed microorganism, say, Aspergillus fumigatus (black mould), appears in the platform. However, until someone needs it, it will not occupy any significant resources in the system. When used for the first time, this reference genome file will be initialised with correct data. A file's initialisation can be a complex computational task, say, the computation of a custom quality metric, or mapping short reads to a genome. This feature opens a number of interesting possibilities for the system as you will see when you start using the platform!Data Management
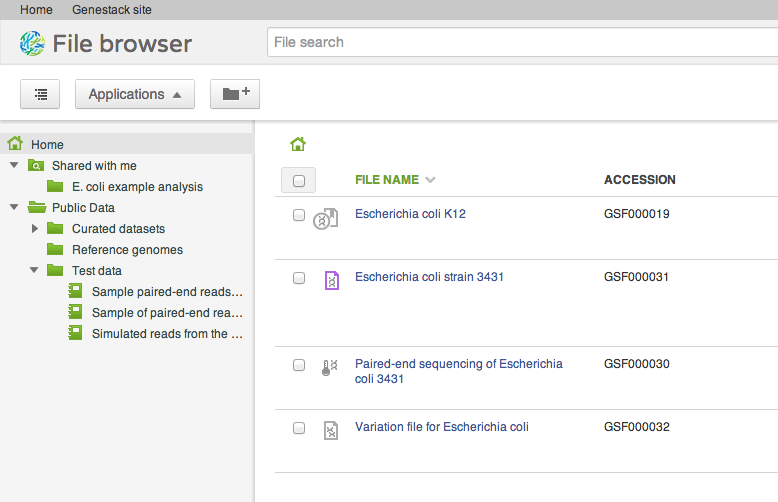
After signing into the system, you will find yourself viewing your files. Every user has a home folder for their own files, a collection of folders shared with user groups (see below), and a connection to public datasets in the Public Data folder.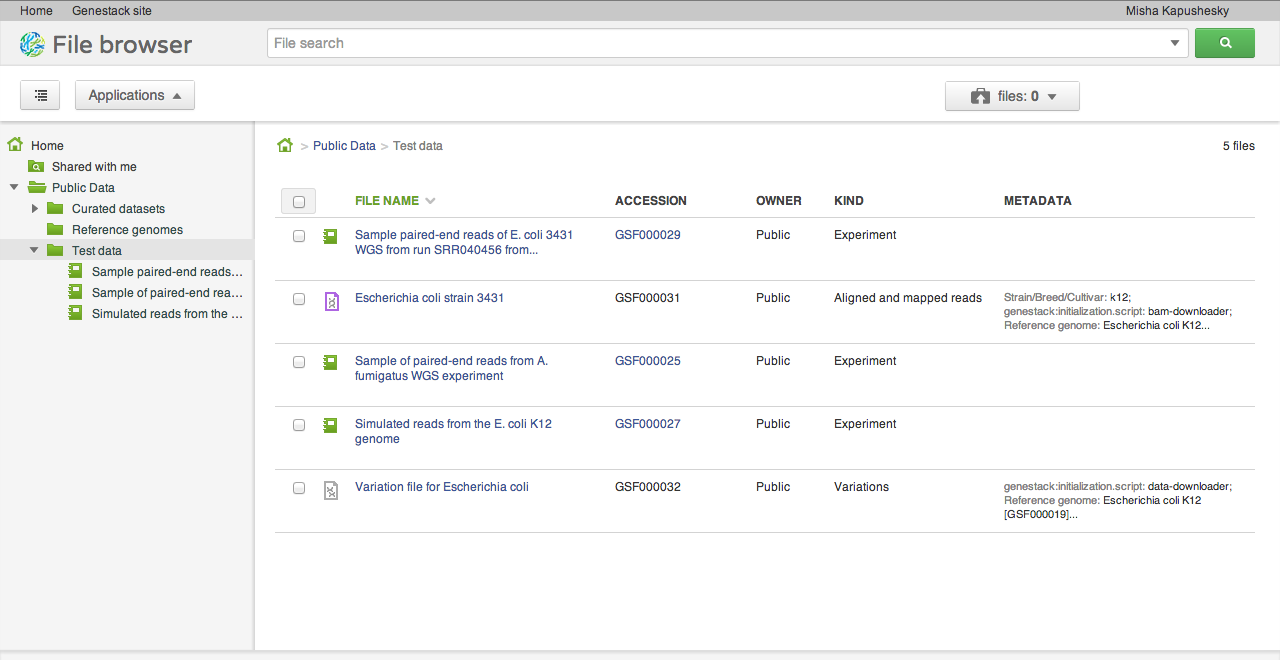
Applications
 For every file or a selection of files the system lists applications that can be used to open the selection. There are two places where applications appear: in the applications pane at the top of the file browser, and in the context menu that shows up on clicking a file name.
For every file or a selection of files the system lists applications that can be used to open the selection. There are two places where applications appear: in the applications pane at the top of the file browser, and in the context menu that shows up on clicking a file name. Copying files
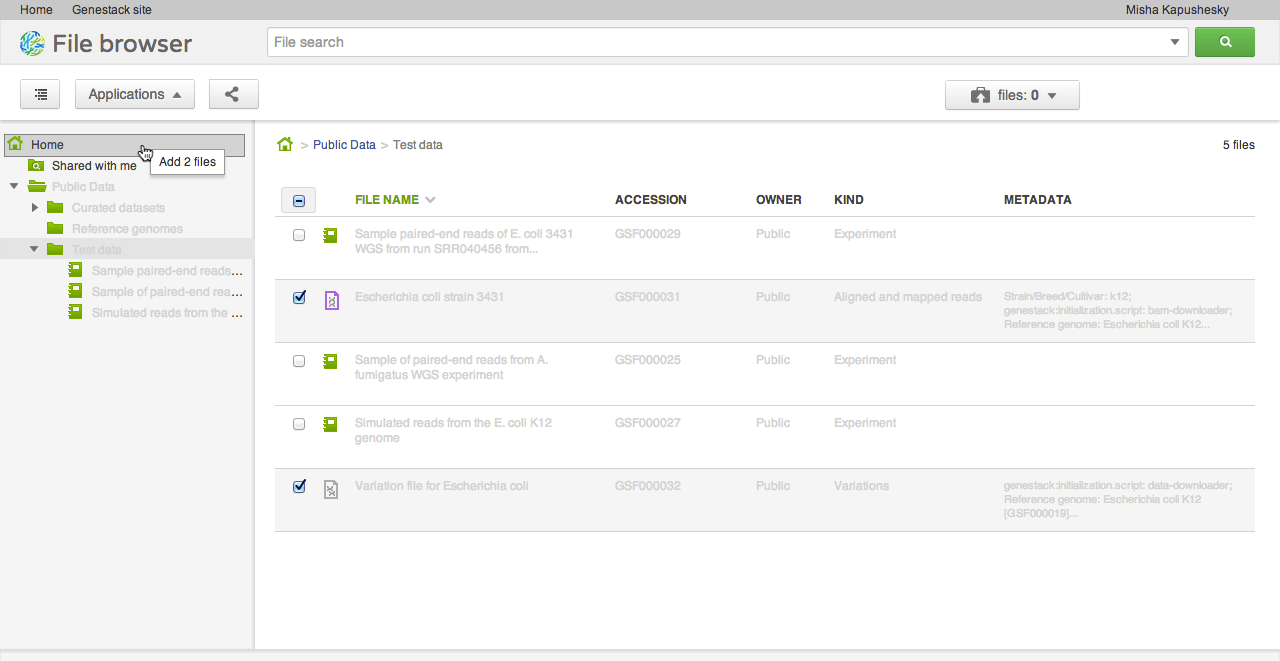 Our file system is different from what you might be used to in one important aspect: files can appear in multiple folders simultaneously (in Unix this feature is known as a hard link). To add one or several files to a folder, drag your selected file(s) on top of the folder where you want to add them. This does not copy the data, and is a quick, cheap operation: it makes the selected files appear in the destination directory.
Our file system is different from what you might be used to in one important aspect: files can appear in multiple folders simultaneously (in Unix this feature is known as a hard link). To add one or several files to a folder, drag your selected file(s) on top of the folder where you want to add them. This does not copy the data, and is a quick, cheap operation: it makes the selected files appear in the destination directory. Briefcase 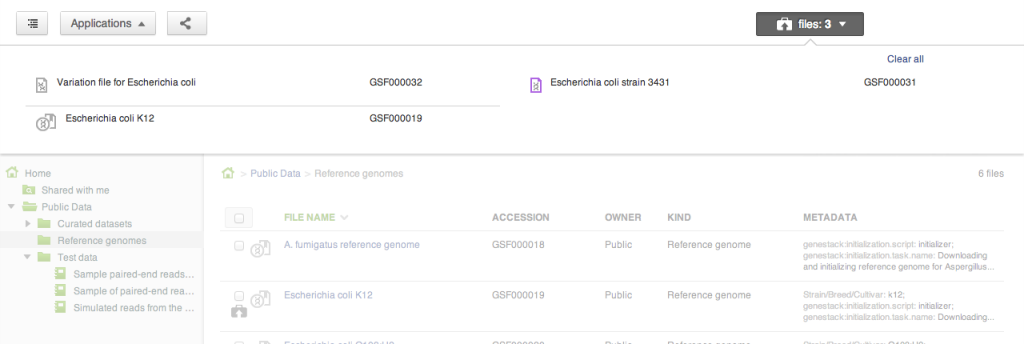
Sometimes you want to collect a bunch of files from different places in the file system and do something with them as a collection: perhaps you want to add them to a new folder, or open them all in one application (say, the Genome Browser, or the Short Read Aligner). Briefcase allows you to add files one at a time to the collection using the briefcase icon next to the file name, and then work on them all together when the briefcase is open. 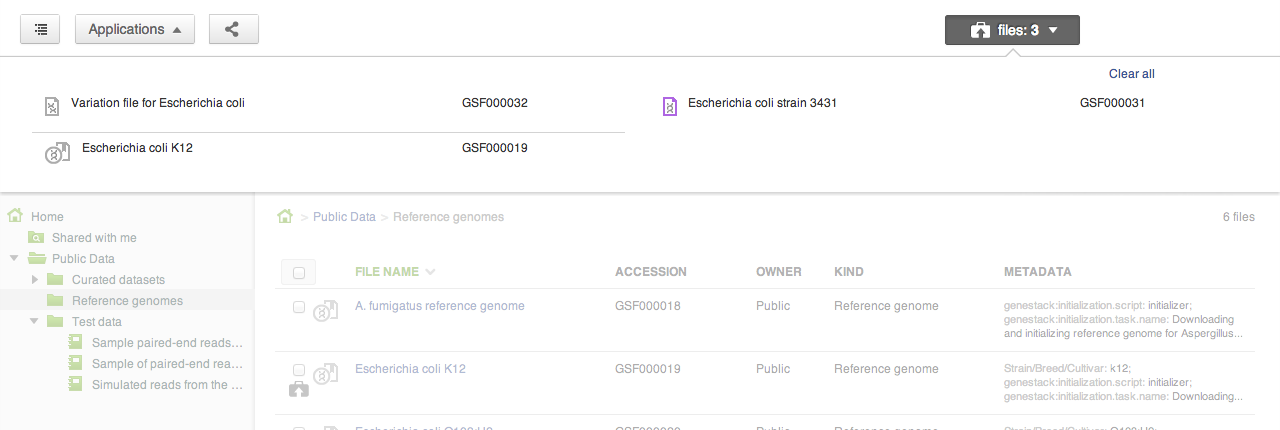
Searching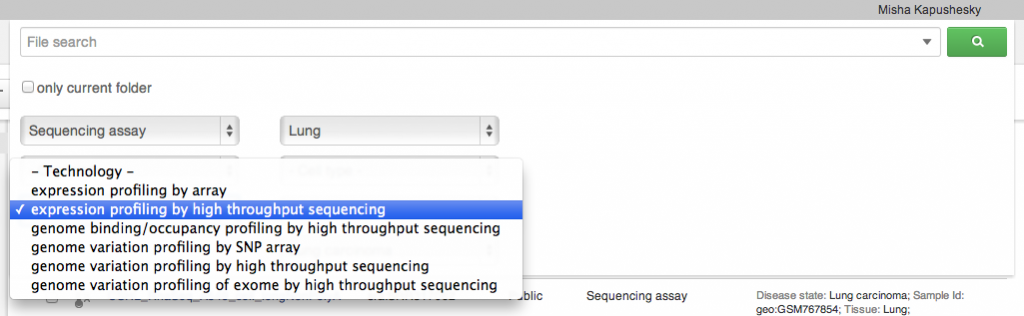
We built a simple but powerful search engine into the system. It can search by file type, technology, organism and a number of metadata attributes, such as tissue, cell type or cell line, and disease. As always, you can run your applications on any result set that you see on the screen. The search is done on private, shared and public datasets, but can be restricted to the folder that you are currently in, if necessary (extending down to its subfolders). 
Sharing and Collaboration
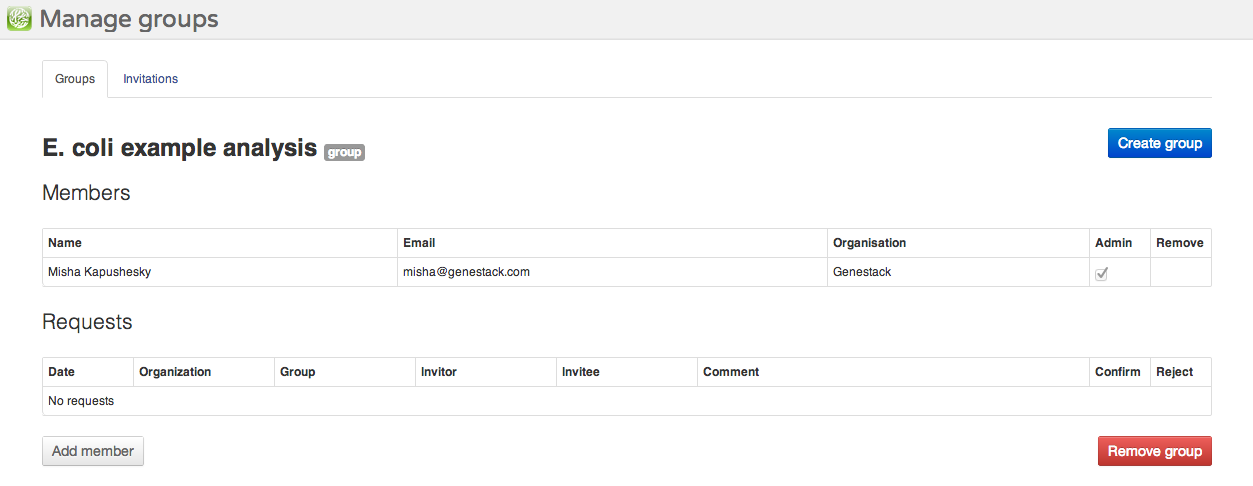
 To share your data and results, you can create groups. A group is represented by a folder to which multiple users are granted access with different permissions. To create a group, select Manage groups from the user menu in the top right corner of the screen. All Genestack Platform users can create as many groups as they want, all you need is to name your new group. In the Manage groups screen, you can create a new group and manage its members.
To share your data and results, you can create groups. A group is represented by a folder to which multiple users are granted access with different permissions. To create a group, select Manage groups from the user menu in the top right corner of the screen. All Genestack Platform users can create as many groups as they want, all you need is to name your new group. In the Manage groups screen, you can create a new group and manage its members. 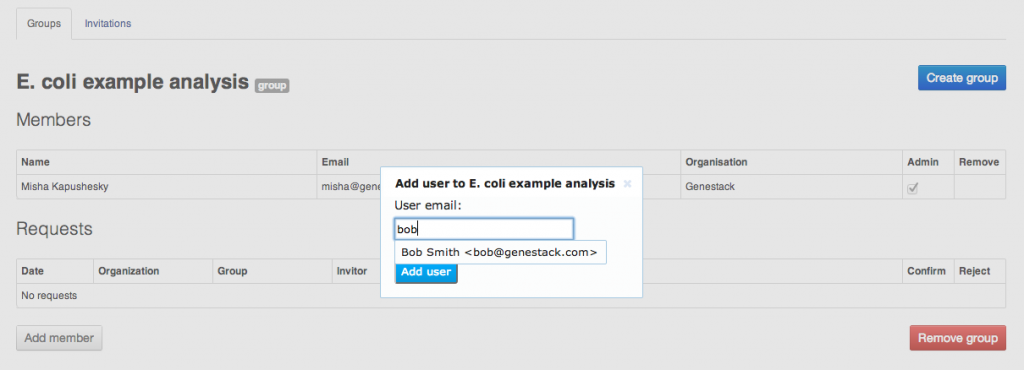
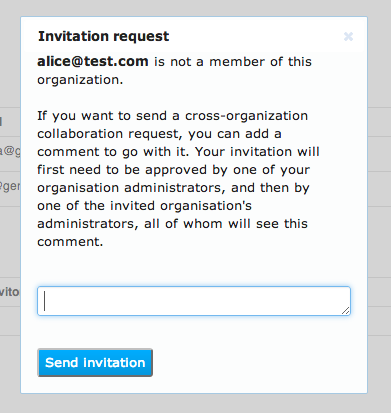 Your organisation's administrator and then the other user's organisation administrator will both need to approve this collaboration request.
Your organisation's administrator and then the other user's organisation administrator will both need to approve this collaboration request.
{kind=link}
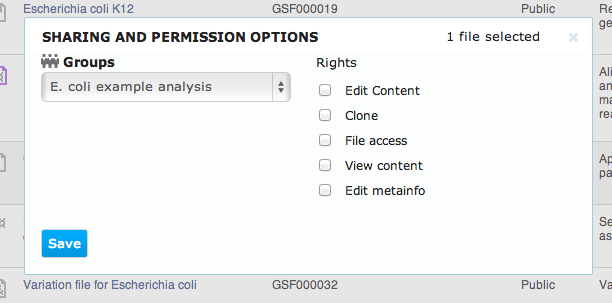
- Edit content - allows users to modify the content of the file
- Clone - allows users to create a full copy of the file in their own private space
- File access - allows users to see the file when listing shared folder contents, i.e. to know about the file's existence
- View content - allows users to view the file content
- Edit metainfo - allows users to modify the file's meta-information attributes
Public Datasets
Our alpha release does not yet expose the capacity to load your own data. To help you get started and play around with the system, we loaded a few interesting public datasets. They are located in the Public Data folder and include a selection of data from the ENCODE project, the Illumina Body Map dataset from ArrayExpress at the EBI, and a few others: whole genome sequencing, RNA-seq, ChIP-seq of human, model organism and microbial genomes. We'll explain in a separate post how we organise and release public datasets. They are stored and are available free of charge for all our users.Applications
Genestack Platform is designed to support developing, hosting and deploying different kinds of applications, both computational ones with long running times and interactive data visualisation apps. We developed several apps to help get you started analysing data. Here's an overview of these apps and of some of their unique features.Genome Browser
Clearly any self-respecting genomics applications platform needs its own genome browser. We built it to satisfy the following requirements:- Easy sharing of genome views
- Fast performance on tens to hundreds of tracks
- Prefer simplicity over data clutter
- Customisable visualisations
Launching the Genome Browser
Genome Browser can open files of the following types:- Reference Genome
- Sequencing Assay
- Aligned and Mapped Reads
- Variations
Genome Browser Interface
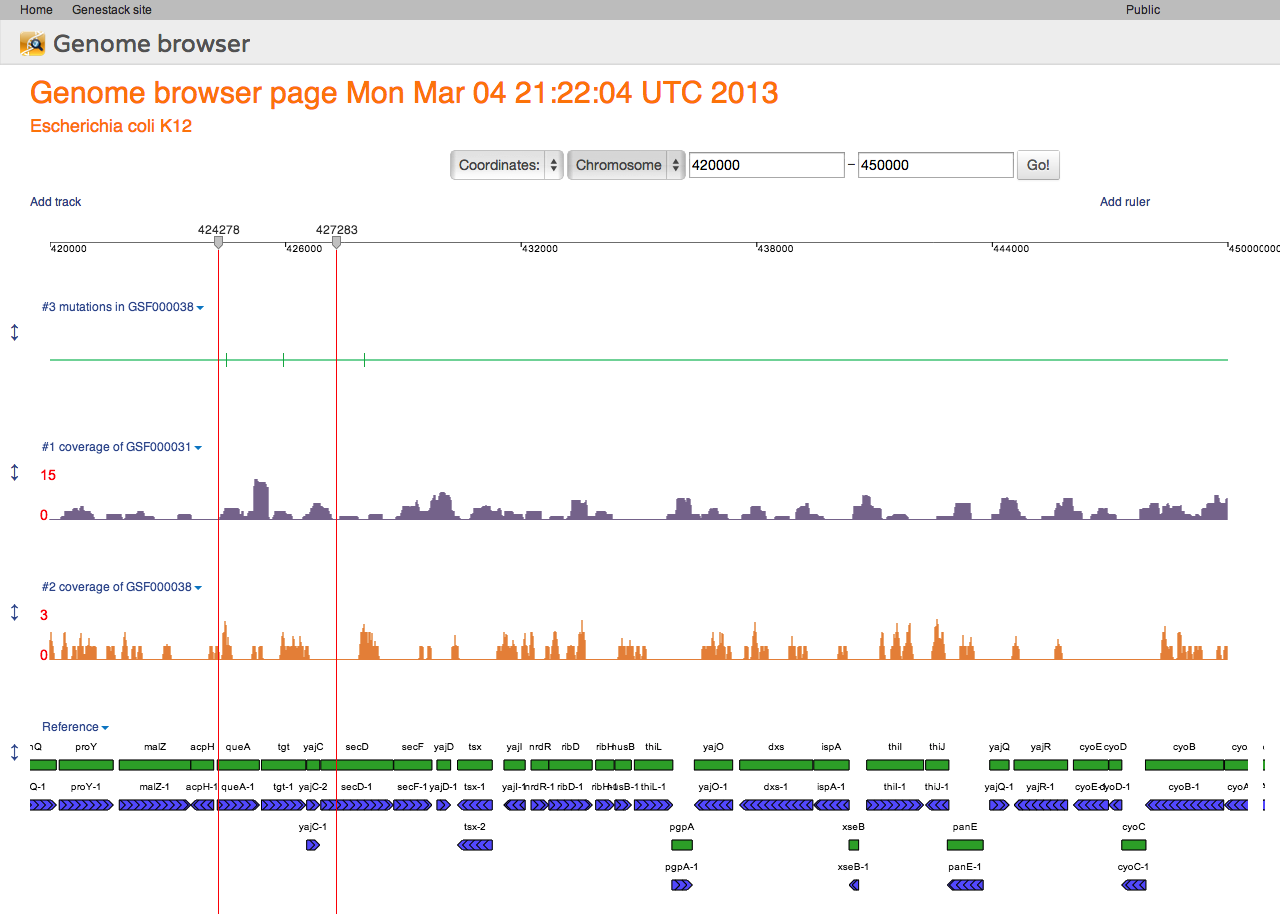
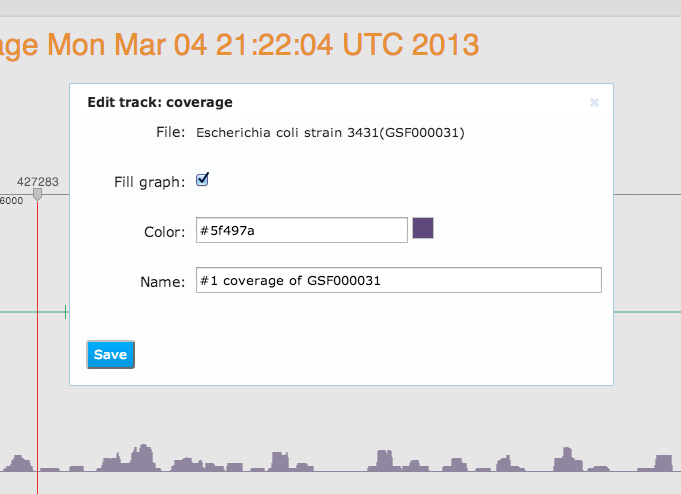
Genome Browser Pages are Files
Following our core idea that everything is a file, every genome browser page is a file, too! When you first open a reference genome, a new file is automatically created for you in your home folder. This file is a view into that specific reference genome. Anything you do in that view will be saved instantly.
Try it out, and watch out for future posts, going into more detail about the genome browser. We'll introduce ruler markers, mathematical formula tracks, and other exciting features.
Quality Control
A simple quality control application for sequencing assays is provided. You can run it on Sequencing assay files and also on Aligned and mapped reads files. It outputs a number of basic QC metrics, such as the average base quality from the start of the read, GC content, uncalled bases, coverage depth and so forth.
In almost all cases, the application makes the best effort to start producing a visual response as soon as it has read a chunk of the data, so that you will get some information about your data quality nearly right away.
Short Read Aligner
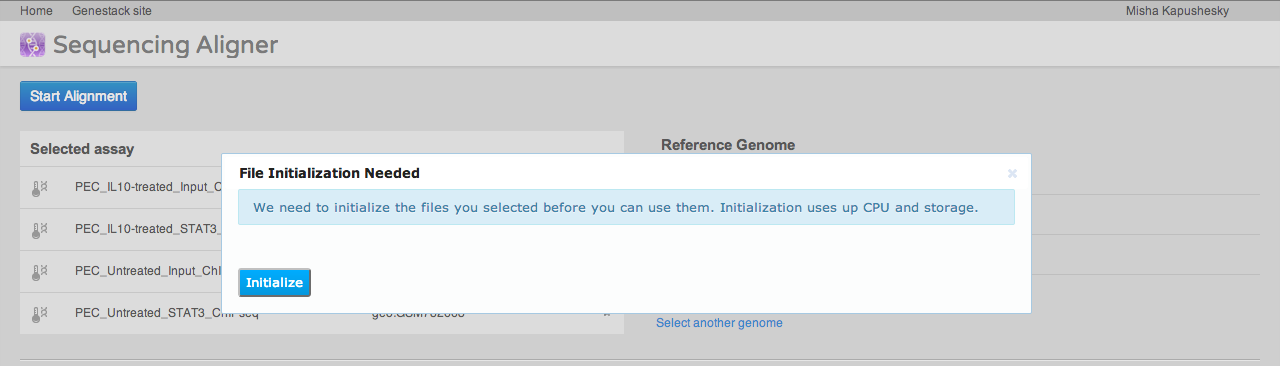
A key step of many high-throughput sequencing workflows is to align short reads to a reference genome. Our short read aligner integrates several popular aligners, namely bwa, bowtie2 and TopHat. It can be called on one or many Sequencing assay files and has a simple interface: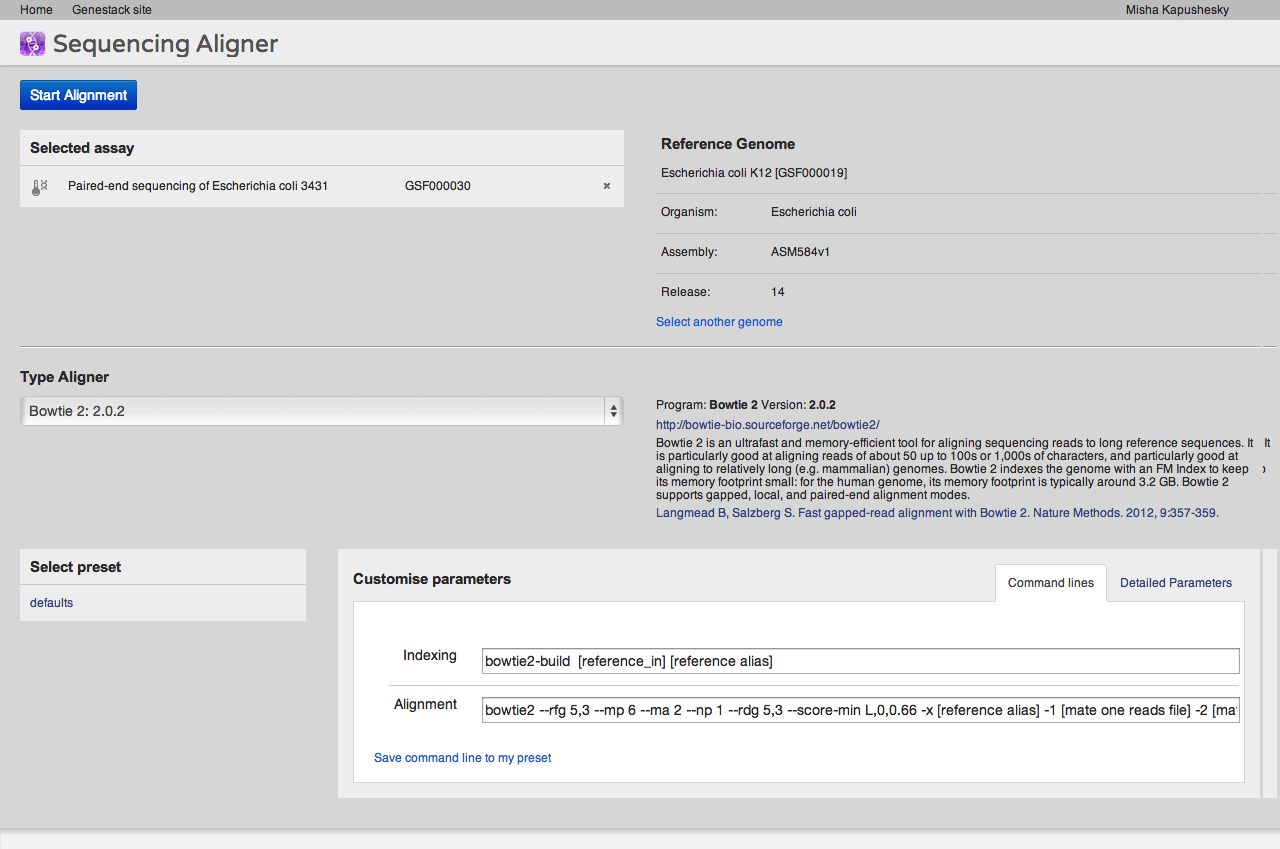
The aligner will process multiple assays, distributing them to our compute cluster as necessary. If you select an uninitialised sequencing assay or try to align to an uninitialised reference genome, the system will ask you if you want to initialise these files first. As described above, initialisation is usually a separate process, and may involve loading data and/or performing computations, in other words, using up storage and CPU.