Genestack sponsored Genome Informatics 2014 (GI2014), a four days conference held at Churchill College, in Cambridge. We were very excited to attend such an established event, and for many of us this 14th edition was the first. Needless to say, our expectations were met by four days packed with exciting talks about Genomics, Sequencing and Bioinformatics, with topics going from the future of clinical genomics to the latest advancements in compression algorithms. Read on to see day-by-day summaries of the conference!
Day 1, Session 1: Personal and Medical Genomics
 GI2014 has been a precious chance for Genestack's bioinformatics team to meet our users in person, giving demos of the platform and introducing researchers and sequencing users to their free Genestack accounts. We showcased our newest apps for differential gene expression and single-cell RNA-seq analysis, and new users were shown how to use our powerful Data Flow Editor to easily reproduce their custom pipeline using one of the 1600 public datasets on Genestack. The first day's talks were not less impressive: Atul Butte broke the ice discussing the risk of using sequencing technology to investigate traits and genotypes separately while ignoring the environmental factors, possibly ending up duplicating a sequencing experiment. Yaoqing Shen discussed the Personalised Oncogenomics project (POG), describing a highly integrated workflow which leverages both RNA-seq and Whole Genome Sequencing. She showed examples of using the supported decision-making approach for people suffering from incurable malignancies. This approach seems to have great potential to improve patient care.
GI2014 has been a precious chance for Genestack's bioinformatics team to meet our users in person, giving demos of the platform and introducing researchers and sequencing users to their free Genestack accounts. We showcased our newest apps for differential gene expression and single-cell RNA-seq analysis, and new users were shown how to use our powerful Data Flow Editor to easily reproduce their custom pipeline using one of the 1600 public datasets on Genestack. The first day's talks were not less impressive: Atul Butte broke the ice discussing the risk of using sequencing technology to investigate traits and genotypes separately while ignoring the environmental factors, possibly ending up duplicating a sequencing experiment. Yaoqing Shen discussed the Personalised Oncogenomics project (POG), describing a highly integrated workflow which leverages both RNA-seq and Whole Genome Sequencing. She showed examples of using the supported decision-making approach for people suffering from incurable malignancies. This approach seems to have great potential to improve patient care.  The third speaker, Orion Buske, introduced PhenomeCentral, a repository aimed at improving rare disease identification by finding of phenotypic similarity between patients and identifying novel phenotypic clusters. One of the highlights was stressing the difficulties in acquiring standardised curated metadata from clinicians, as different professionals might describe the same patient with different terms.
The third speaker, Orion Buske, introduced PhenomeCentral, a repository aimed at improving rare disease identification by finding of phenotypic similarity between patients and identifying novel phenotypic clusters. One of the highlights was stressing the difficulties in acquiring standardised curated metadata from clinicians, as different professionals might describe the same patient with different terms.  Konrad Karczewski demonstrated a new open-source tool, LOFTEE (Loss Of Function Transcript Effect Estimator) for annotation of loss-of-function variants. This tool was applied to a large scale dataset or almost a hundred thousand genomes, including samples from Finland and UK, to characterize the landscape of knockouts (homozygous loss-of-function (LoF) variants) in humans. Liz Worthey discussed the benefits of Whole Genome Sequencing (WGS) over Whole Exome Sequencing (WES) technologies in the context of clinical diagnosis. With current technologies, WGS does not appear to be significantly more expensive than WES, while detecting around 4% uncalled variants from WES. Quite impressively, Worthey and her collaborators are working on a platform which is able to provide a clinical report within a couple of days from sample delivery.
Konrad Karczewski demonstrated a new open-source tool, LOFTEE (Loss Of Function Transcript Effect Estimator) for annotation of loss-of-function variants. This tool was applied to a large scale dataset or almost a hundred thousand genomes, including samples from Finland and UK, to characterize the landscape of knockouts (homozygous loss-of-function (LoF) variants) in humans. Liz Worthey discussed the benefits of Whole Genome Sequencing (WGS) over Whole Exome Sequencing (WES) technologies in the context of clinical diagnosis. With current technologies, WGS does not appear to be significantly more expensive than WES, while detecting around 4% uncalled variants from WES. Quite impressively, Worthey and her collaborators are working on a platform which is able to provide a clinical report within a couple of days from sample delivery.  Other speakers for the first day included Daryanaz Dargahi, who described a pipeline for identifying splice variants used to study oncogenesis; Atul Sethi, who described an intergrated analysis leveraging data from The Cancer Genome Atlas (TCGA) to identify biomarkers of ovarian cancer; and David McCarthy, with an application of WGS and Linear Mixed Models (LMMs) to identify the contribution of different classes of genetic variants to type 2 diabetes.
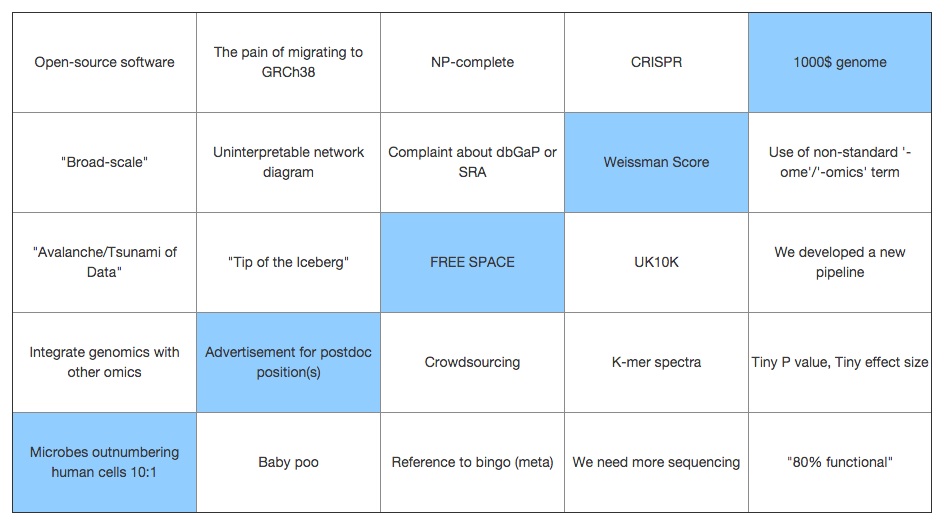
Other speakers for the first day included Daryanaz Dargahi, who described a pipeline for identifying splice variants used to study oncogenesis; Atul Sethi, who described an intergrated analysis leveraging data from The Cancer Genome Atlas (TCGA) to identify biomarkers of ovarian cancer; and David McCarthy, with an application of WGS and Linear Mixed Models (LMMs) to identify the contribution of different classes of genetic variants to type 2 diabetes.  The first day saw a few more innovations. After 24 years of Powerpoint, GI2014 treated us to the official conference bingo. The GI2014 card contained classics like "NP-complete" and "we need more sequencing", joined by new entries like "the pain of migrating to GRSh38″ and "UK10K". The initiative was well received, with both speakers and attendees joining the hunt for jargon.
The first day saw a few more innovations. After 24 years of Powerpoint, GI2014 treated us to the official conference bingo. The GI2014 card contained classics like "NP-complete" and "we need more sequencing", joined by new entries like "the pain of migrating to GRSh38″ and "UK10K". The initiative was well received, with both speakers and attendees joining the hunt for jargon.