 It performs quality control checks on raw sequencing data and generates a bunch of QC reports telling you in which areas there may be problems. These reports cover the following:
It performs quality control checks on raw sequencing data and generates a bunch of QC reports telling you in which areas there may be problems. These reports cover the following: - basic statistics
- sequence length distribution
- per sequence GC content
- per sequence quality scores
- per base sequence content
- sequence duplication level
- overrepresented sequences
We'll show you examples of the above using FastQC reports prepared for data from 2013 publication by Hibaoui et al. on early developmental transcriptional signature of Down syndrome. This experiment (along with hundreds of thousands of others) is available on the Genestack in our Public Experiments Index. The reports can be found here. Let's go through all the reports generted by the FastQC app: Basic Statistics report tells you about the basic stats of your data: reads type, number of reads, GC content and total sequence length. Sequence length distribution report tells you about whether or not your sequences have the same length.
Sequence length distribution report tells you about whether or not your sequences have the same length. Per sequence GC content graph will help you access whether your data is contaminated. In a random library you should expect a roughly normal GC content distribution. Looking at the type of peaks on the graph (sharp/broad) will give you hints on the types of contaminants in your data set.
Per sequence GC content graph will help you access whether your data is contaminated. In a random library you should expect a roughly normal GC content distribution. Looking at the type of peaks on the graph (sharp/broad) will give you hints on the types of contaminants in your data set.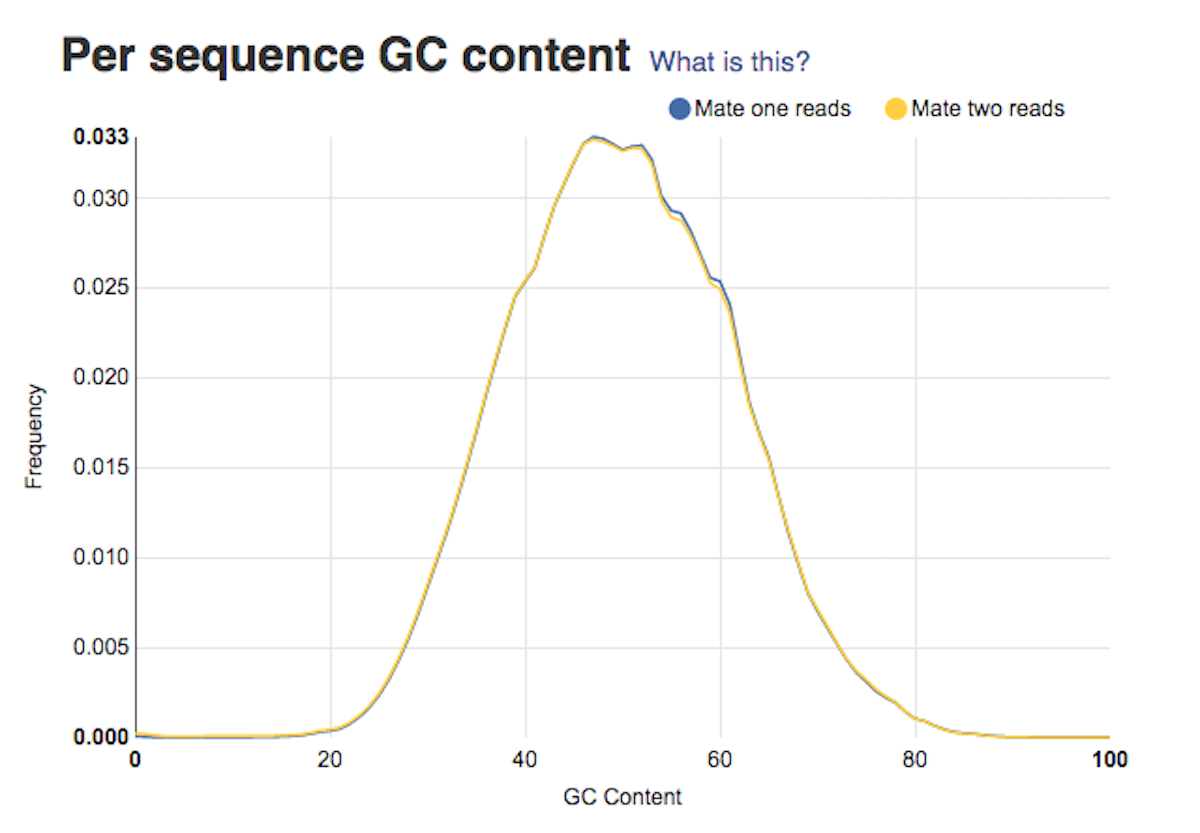 Per base sequence quality plots show a range of quality scores for each position in the reads. A good sample will have qualities all above 28. This report will help you decide if your data needs trimming - e.g. in a situation where the quality of the reads degrades over time.
Per base sequence quality plots show a range of quality scores for each position in the reads. A good sample will have qualities all above 28. This report will help you decide if your data needs trimming - e.g. in a situation where the quality of the reads degrades over time.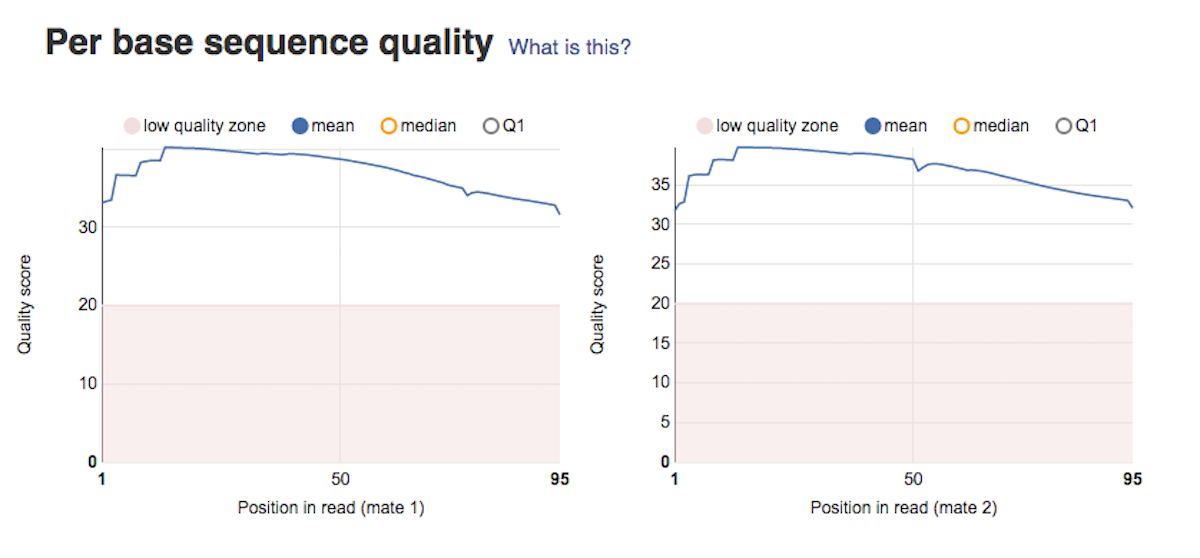 Per sequence quality score plot shows the frequencies of quality scores in a sample. If the reads are of good quality, the peak on the plot should be shifted to the right as far as possible. Errors seen in this report warn you about a general loss of quality of your raw reads within a run.
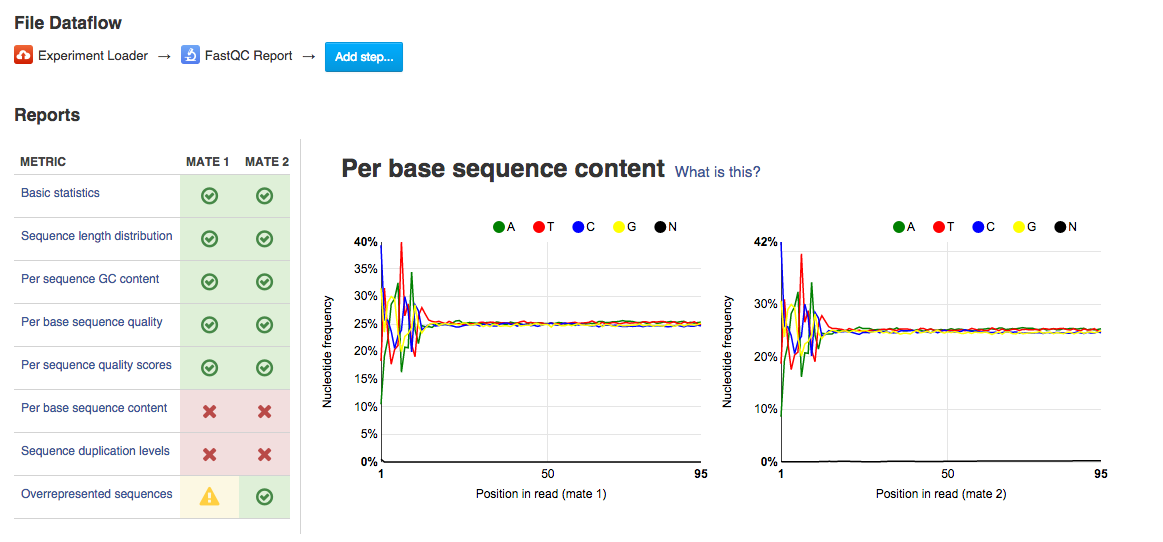
Per sequence quality score plot shows the frequencies of quality scores in a sample. If the reads are of good quality, the peak on the plot should be shifted to the right as far as possible. Errors seen in this report warn you about a general loss of quality of your raw reads within a run.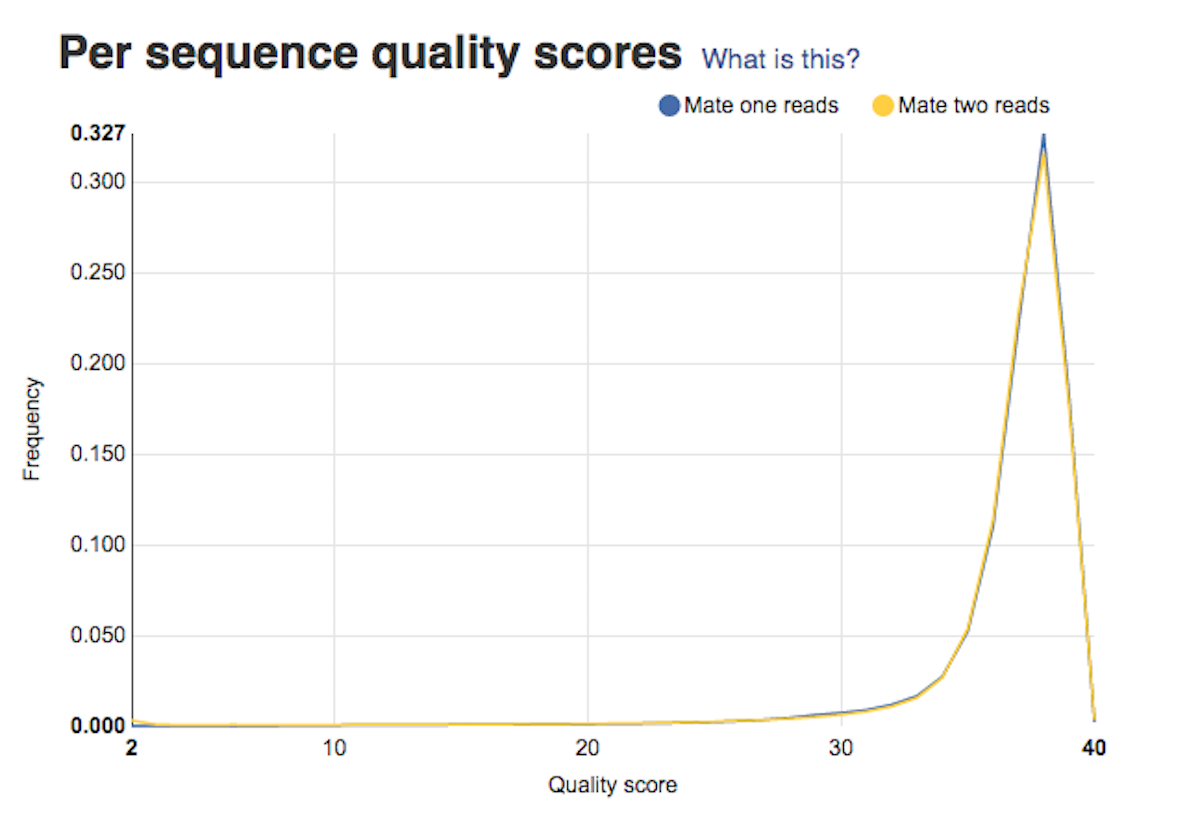 Per base sequence content plot shows the four nucleotides' proportions for each position. In a random library you expect no nucleotide bias and the lines should be almost parallel with each other. A common problem that can be discovered looking at this graph is a presence of overrepresented sequences in the sample.On the picture belowe we can see a bias at the beginning of the reads, which is common for RNA-Seq data.
Per base sequence content plot shows the four nucleotides' proportions for each position. In a random library you expect no nucleotide bias and the lines should be almost parallel with each other. A common problem that can be discovered looking at this graph is a presence of overrepresented sequences in the sample.On the picture belowe we can see a bias at the beginning of the reads, which is common for RNA-Seq data.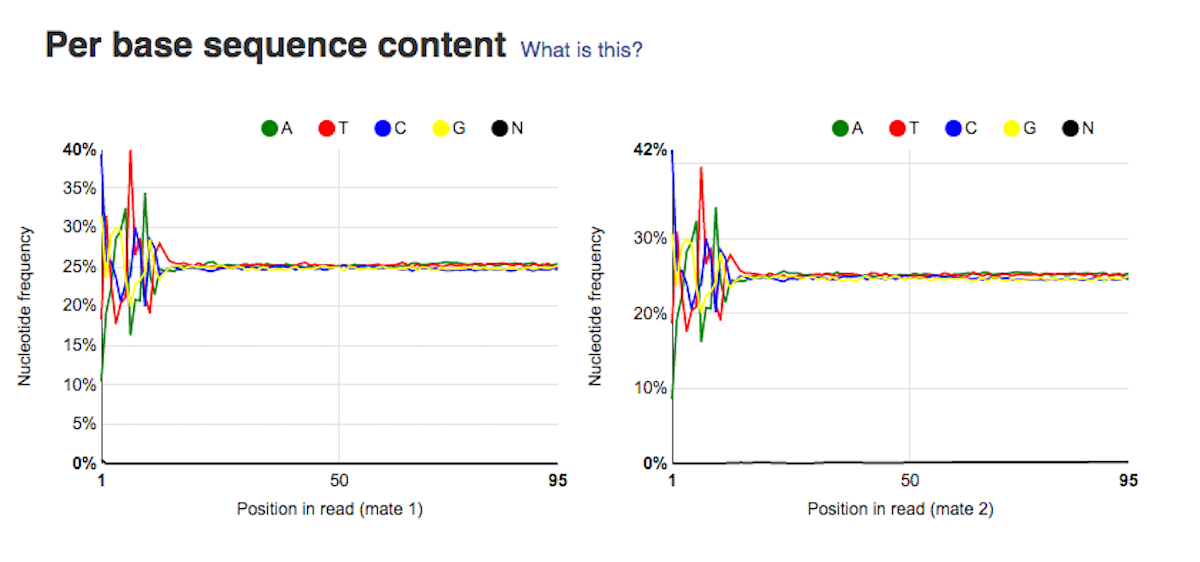 Sequence duplication level graph shows the proportion of the library made up of sequences with different duplication levels. Sequences with 1, 2, 3, 4, etc duplicates are grouped to give the overall duplication level. In the example below we can see that in this sample, 15% of sequences are duplicated 10 times, 7% are duplicated 100 times and so on. Generally, there are two potential types of duplicates in a library-technical or biological with both being shown on this graph.
Sequence duplication level graph shows the proportion of the library made up of sequences with different duplication levels. Sequences with 1, 2, 3, 4, etc duplicates are grouped to give the overall duplication level. In the example below we can see that in this sample, 15% of sequences are duplicated 10 times, 7% are duplicated 100 times and so on. Generally, there are two potential types of duplicates in a library-technical or biological with both being shown on this graph.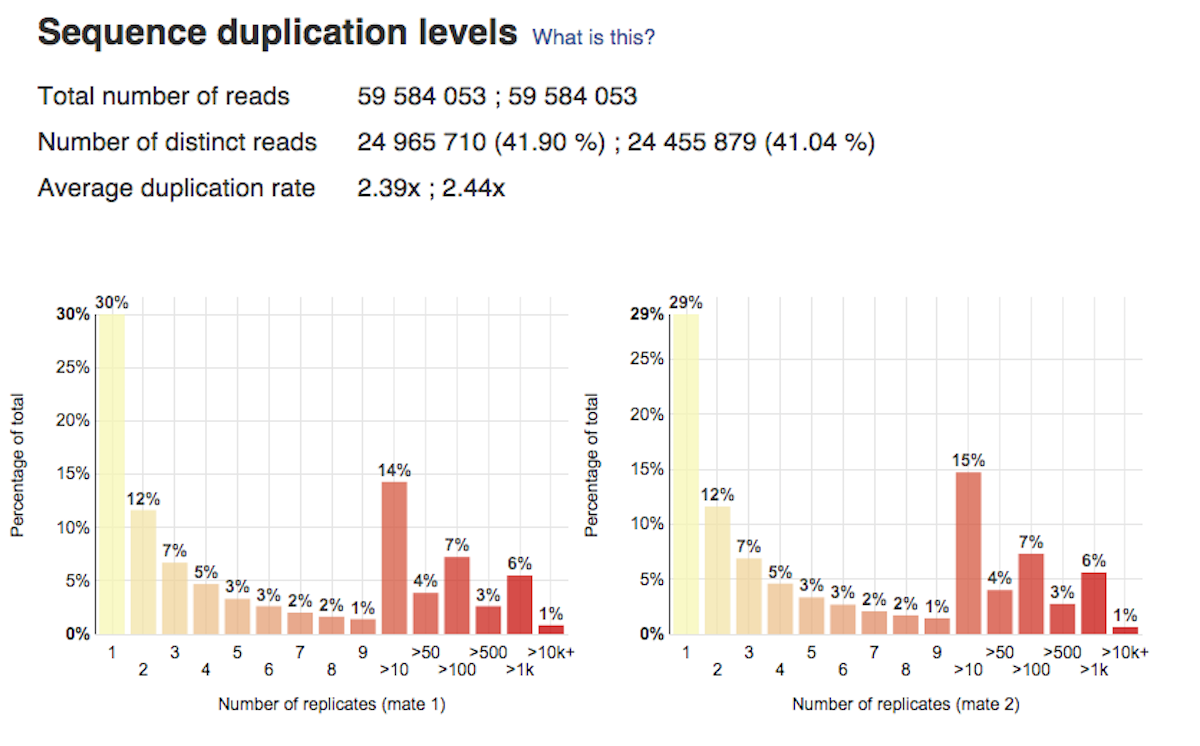 Last, but not least, the Overrepresented Sequences report will warn you if any of the sequences makes up more than 0.1% of the entire sample.
Last, but not least, the Overrepresented Sequences report will warn you if any of the sequences makes up more than 0.1% of the entire sample. 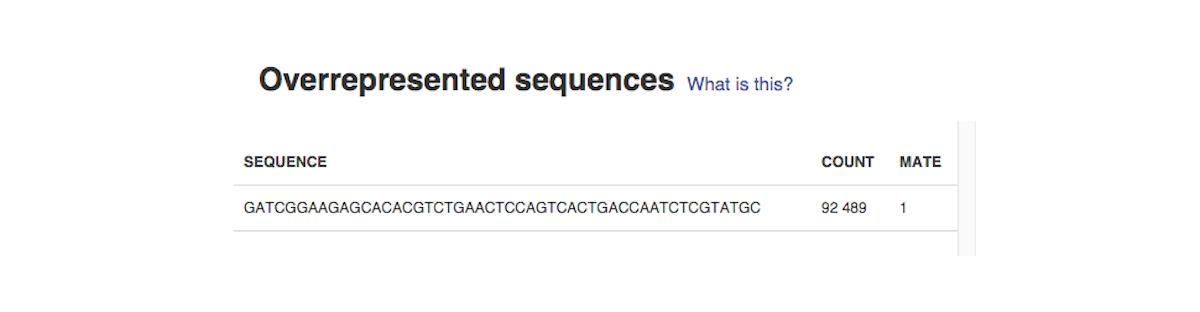 The app is based on FastQC, developed by Simon Andrews and his colleagues at the Babraham Institute (Cambridge, UK). All of these reports will later help you decide if your data needs preprocessing, and if so, what type of preprocessing. After you run several FastQC reports (which you could do using our QC dataflow that was discussed in the Differential Gene Expression Analysis tutorial), you could compare them using our Multiple QC Report app.
The app is based on FastQC, developed by Simon Andrews and his colleagues at the Babraham Institute (Cambridge, UK). All of these reports will later help you decide if your data needs preprocessing, and if so, what type of preprocessing. After you run several FastQC reports (which you could do using our QC dataflow that was discussed in the Differential Gene Expression Analysis tutorial), you could compare them using our Multiple QC Report app. 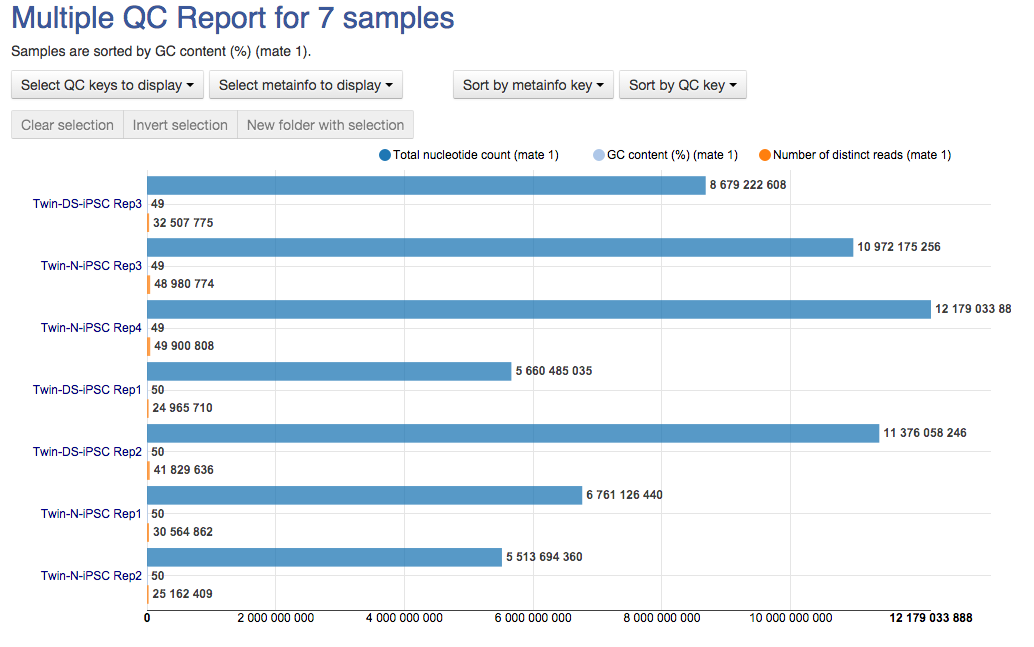 We hope you'll enjoy our new app. Let us know what you think about it, Genestack team
We hope you'll enjoy our new app. Let us know what you think about it, Genestack team