Introduction
Multi-omics research has emerged as a powerful approach to gain comprehensive insights into various biological processes by integrating different types of data from various molecular levels. This strategy enables researchers to examine the same biological phenomenon through multiple lenses, thereby providing a more comprehensive understanding of the underlying mechanisms. However, such an approach entails generating extensive and diverse data sets compared to conventional genomic or transcriptome analyses. Furthermore, as different types of data are typically generated by distinct laboratories or organisations, harmonising and integrating them poses a significant challenge.
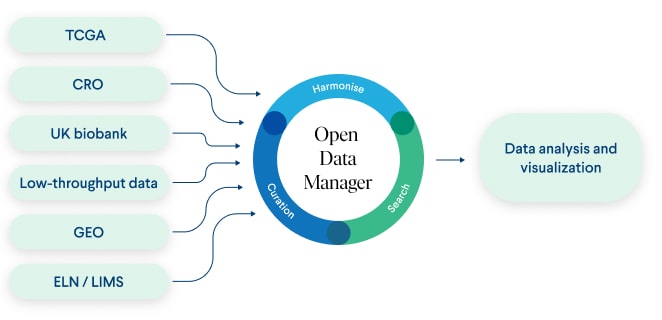 Figure 1. The schematic image of data flow in the Genestack platform.
Figure 1. The schematic image of data flow in the Genestack platform.
Data volumes are also a challenge in the life sciences industry. Our customers conducting multi-omics research on cancer data use hundreds and thousands of datasets for a single analysis, sourced from varying origins. Genestack platform serves as a solution to effectively collect, store , consolidate and harmonise such data. Beyond working with inhouse data, our platform enables data upload from external sources, e.g. data produced by collaborators and contract research organisations (CROs), or produced by stand-alone platforms and ELNs/LIMS, facilitates the inclusion of public data and supports high- and low-throughput analysis results. These tasks are made possible through our robust and dependable system, which acts as a single point of truth. Such a system is especially vital when conducting multi-omics research, which requires large amounts of data sourced from diverse origins. In the current study, we aimed to elucidate the workflow of our clients using publicly available data and to demonstrate how our data management platform can aid in optimising the analysis of multi-omics data. With Genestack ODM, true multi-omics analysis is not only feasible but also simplified allowing you to combine both low and high complexity data in a single point of truth.
Challenges in Multi-Omics Research
Another challenge is data access. Mere availability of data is not enough if it cannot be accessed from anywhere and anytime. The Genestack platform offers easy access through the browser and supports login via SSO, which eliminates the need to store data on a local computer. This enables researchers to retrieve data efficiently and seamlessly.
Moreover, the importance of clinical and phenotypic data (often referred simply as metadata) tracking in data analysis cannot be overstated. Researchers can gain additional insights into biological processes by interrogating expressions and variants based on metadata, which can be used for "interpretation using additional covariates." This technique can help identify new targets for drug development and shed light on disease mechanisms, such as cancer. Therefore, proper metadata tracking is crucial for successful multi-omics analysis.
Experimental Design:
As typical large scale multi-omics research requires a large amount of proprietary data with limited access we decided to illustrate the power of harmonised data on using an iconic publicly available dataset. The entire workflow could be easily extrapolated to a large collection of harmonised data available at a typical Life Sciences organisation.
The current investigation employed publicly available TCGA 1 data on Kidney papillary cell carcinoma that was obtained from the xena browser portal 2. The objective of this study was to evaluate gene expression utilising RNA-seq, mutations employing exome-seq, and copy number variations utilising genotyping arrays. The purpose of this evaluation was to determine the differences between the tumours and test whether two tumour types can be distinguished using multi-omics data.
Methodology:
The tools employed in this investigation are common among bioinformaticians and included the R programming language along with the "FactoMineR" package for multi-factor analysis 3 and "pheatmap" package for heatmap visualisation 4. Additionally, we utilised the Genestack R packages to retrieve data directly from the system. It allows one to get data without the need of manually getting it from the system and saves extra time for a researcher.
All data underwent importation into the system utilising a Python script from Xena Browser and harmonisation. The curation tool offered by the Genestack platform facilitated this procedure with minimal time consumption. Metadata templates that had scientific ontologies linked to them provided a robust basis for successful data integration.
Traditionally, data analysis involves the exportation and uploading of data to a local computer. However, our platform provides Application Programming Interfaces (APIs) that enable direct access to the data without the need for exporting. R packages and Python libraries are also available and do not require any API experience. With the aid of a function call, we were able to execute a search for all RNA-seq, exome-seq, and copy number variation data for TCGA1 kidney cancer across all datasets present in the platform. This search was executed directly in the Jupyter notebook and produced data that was easily analysed.
Data Processing and Analysis:
The collected data was imported into the system and underwent harmonisation utilising the tool provided by the Genestack platform. This process was executed expeditiously, owing to the tool's efficient performance. Metadata templates, equipped with scientific ontologies, formed a robust basis for successful data integration. We refer readers to our previous article, "Agriscience Expression Analysis - In an evening", for a detailed description of the data import and cleaning processes.
The primary objective of the analysis was to ascertain whether there were significant statistical differences between two different tumour types: Type one and type two. The first step entailed performing an exploratory analysis, where each type of data was examined independently. To reduce the amount of information, genes that did not exhibit mutations or expression changes were eliminated, as the primary focus was on biological changes. Figures 2-4 display three heatmaps, each representing an omics data type stratified by tumour type.
The heatmap of the mutation data indicated that the number of mutations in the samples was not substantial, as most cancers displayed significantly more red colour in the heatmap. Distinguishing between the two cancers based on the heatmap of gene expression alone was challenging. Similarly, the heatmap displaying copy number variations failed to distinguish between the two cancer types. However, a multifactor analysis was conducted subsequently to determine if all three types of data combined could effectively differentiate between cancer types.
 Figure 2. The heatmap summarising mutation data for KIRP. The rows are genes and columns are samples, colour represents the number of mutations.
Figure 2. The heatmap summarising mutation data for KIRP. The rows are genes and columns are samples, colour represents the number of mutations.
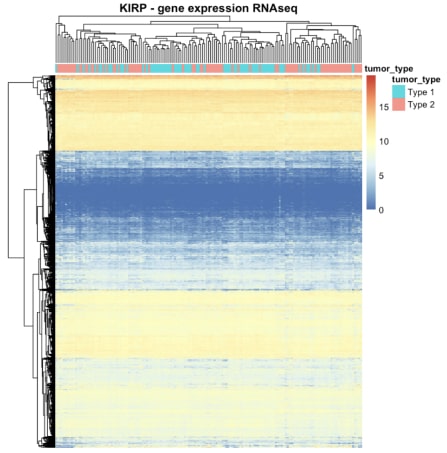 Figure 3. The heatmap summarising RNA-seq gene expression data for KIRP.
Figure 3. The heatmap summarising RNA-seq gene expression data for KIRP.
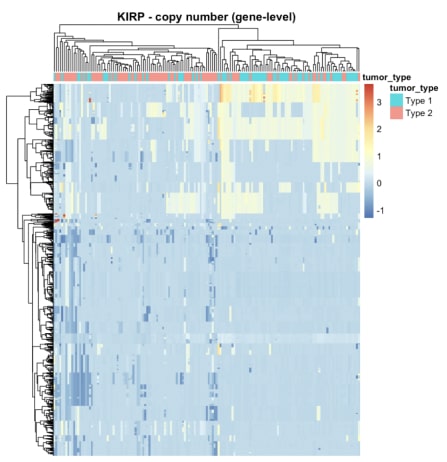 Figure 4. The heatmap of copy number data for KIRP.
Figure 4. The heatmap of copy number data for KIRP.
The presented heatmaps indicate that each omics type contains some discernible signal, yet none of them, in isolation, provides a complete explanation of the cancer subtypes. Instead, each omics type offers only a limited understanding of the distinct characteristics of the tumours when compared to healthy cells.
To gain further insights, we utilised multifactor analysis (MFA), a prevalent approach in matrix factorisation methods that integrates different data types. MFA extends the principle of principal component analysis (PCA) to multiple data domains. In our study, we implemented MFA using the FactoMineR package version 3. The output of MFA is a two-dimensional factorisation of the multi-omics data. By plotting each tumour as a point in a 2D scatter plot, we can evaluate the effectiveness of MFA factors in separating the different cancer subtypes.
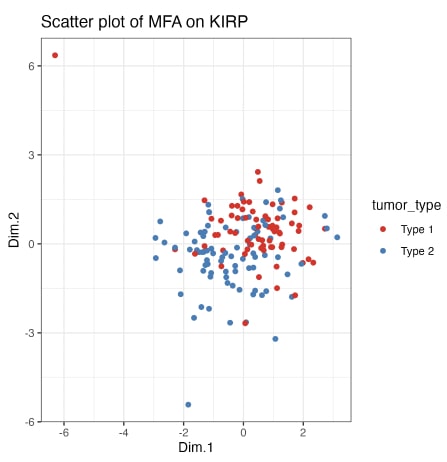 Figure 5. The scatterplot of Multi Factor Analysis results by tumour type.
Figure 5. The scatterplot of Multi Factor Analysis results by tumour type.
As illustrated in Figure 5, the multifactor analysis indicates that there are discernible dissimilarities between the two tumour types, albeit with some degree of overlap. While a definitive classification of tumour types with 100 percent accuracy is not currently feasible, it is evident that type one and type two tumours possess distinctive features. Further investigation is required to corroborate the findings using non-negative matrix factorisation, by exploring the activated signalling pathways through enrichment analysis or by examining the correlation with other relevant variables, such as gender and histological tumour type.
It is noteworthy that the primary objective of this experiment was to conduct an exploratory analysis and assess the efficacy of the Genestack platform in handling multi-omics data, which was deemed successful.
Our findings confirmed previous scientific evidence that green tea contains higher levels of antioxidants than black tea 5, further highlighting its potential health benefits. Furthermore, both tea types were found to contain theanine, an amino acid known for its calming effects on the brain. However, green tea was observed to have higher levels of theanine than black tea.
Conclusion
In summary, this study highlights how the integration of multiple omics datasets using multifactor analysis can reveal meaningful differences between KIRP cancer tumour subtypes. The use of the Genestack platform facilitated the efficient handling of data and allowed the researchers to focus on analysis, resulting in faster and more efficient outcomes.
The ability to conduct a comprehensive analysis in just one evening with limited resources demonstrates the potential of using Genestack for bioinformatics research. Researchers interested in exploring the platform can access a live demonstration or try it themselves by contacting the Genestack team.
Overall, this study showcases the benefits of using advanced bioinformatics tools and platforms in facilitating the analysis of complex datasets, leading to more efficient and insightful results.
For those interested in further exploring we invite you to review our other articles.
Get in touch with us:
info@genestack.com
Request ODM Demo